
William Boisvert, Ph.D.
Professor of Medicine
Center for Cardiovascular Research
John A. Burns School of Medicine
651 Ilalo Street, BSB 311
Honolulu, HI 96813
Email: wab@hawaii.edu
Phone: (808) 692-1567
Research in the Boisvert Lab are focused on exploring immunity and inflammation in the pathogenesis of atherosclerosis.

1. Endothelial cholesterol metabolism
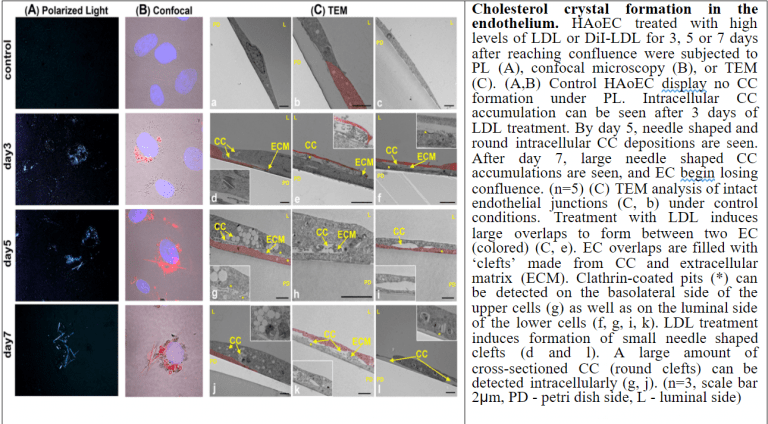
Although much is known about advanced stages of atherosclerosis, events leading to its initiation are poorly understood. This project explores how endothelial cells (EC) behave under hyperlipidemic conditions. We have discovered that when the EC become overwhelmed with lipids they form cholesterol crystals (CC) which are deposited into the so-called subendothelial space. In vivo, we were able to detect the presence of CC in the arterial wall as early as 1 week after high fat diet before any macrophages are detectable. In vitro, we were able to recapitulate the in vivo events by demonstrating the presence of CC on the baso-lateral side of EC when the cells were exposed to physiologic hyperlipidemic conditions. The presence of CC leads to attraction of monocytes to the area and may be the event that initiates the atherogenic process. We are currently pursuing mechanistic studies to determine the LDL uptake process and intracellular metabolism of LDL (specifically, involvement of lipophagy) as well as the role of a highly upregulated ABC transporter called ABCC3 in the basolateral secretion of CC. We are also using a state-of-the-art technique called vessel-on-a-chip to study how precisely altered flow pattern in the vessel leads to changes in the LDL uptake by the endothelium. Moreover, we have an ongoing collaboration with a company in Switzerland that is producing specifically-targeted, drug-filled liposome for us for the express purpose of reducing the CC formation by the endothelial cells and ultimately, lessening the atherosclerotic burden.
2. MicroRNA in cholesterol metabolism and HDL homeostasis

Macrophage foam cell formation is a key feature of atherosclerosis. Recent studies have shown that specific microRNAs (miRs) are regulated in modified low-density lipoprotein (LDL)- treated macrophages, which can affect the cellular cholesterol homeostasis. Undertaking a genome-wide screen of microRNAs regulated in primary macrophages by modified LDL, miR-302a emerged as a potential candidate that may play a key role in macrophage cholesterol homeostasis. We found that transfection of primary macrophages with either miR-302a or anti-miR-302a regulated the expression of ATP-binding cassette (ABC) transporter ABCA1 mRNA and protein. Luciferase reporter assays showed that miR-302a repressed the 3’UTR activity of mouse and human ABCA1. Additionally, transfection of macrophages with miR-302a attenuated cholesterol efflux to apolipoprotein A-1 (apoA-1). In vivo administration of anti-miR-302a to mice with LDL receptor deficiency (Ldlr-/-) fed an atherogenic diet resulted in an increase in circulating plasma HDL levels and a reduction in atherosclerotic plaque size. These studies identify miR-302a as a novel modulator of cholesterol efflux and a potential therapeutic target for suppressing the development of atherosclerosis. We are currently studying how miR302a is regulated and how the anti-miR alters the lipoprotein profile and reverse cholesterol transport capacity. We plan to pursue other microRNAs that show promise in altering HDL levels.
3. Vascular smooth muscle cell migration and fibrous cap formation
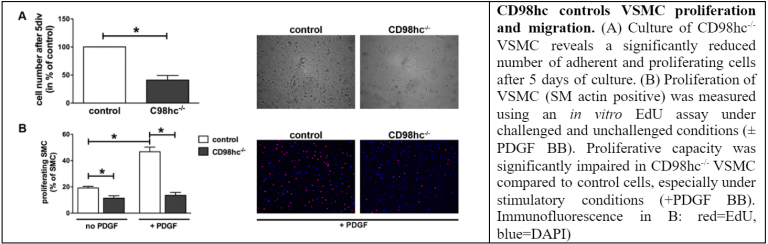
One of the most important events in atherosclerotic plaque formation is vascular smooth muscle cell (VSMC) migration from the media into the intima. This is thought to occur in response to mediators such as cytokines and growth factors from the inflammatory cells in the plaque which cause the VSMC to migrate and become a part of the fibrous cap that forms a protective layer above the necrotic core. CD98 is a transmembrane protein that mediates cell proliferation. We found that CD98 is instrumental in VSMC proliferation and survival and are in the process of studying how CD98 participates in the formation of fibrous caps in atherosclerosis and how CD98 can be manipulated to build and maintain this protective layer of cells and matrix. We have also discovered recently that CD98 may play a very important role in the proliferation of macrophages in the early plaque formation.
4. Gene therapy using hematopoietic stem cells
We are actively pursuing studies on transducing stem cells with genes that have high potential to be anti-inflammatory. Our initial efforts in this area involved therapy with IL-10 utilizing a system in which IL-10 is transduced in the hematopoietic stem cells with the ultimate goal of overexpressing the cytokine in macrophages. IL-10 is a prototypic anti-inflammatory cytokine that we have shown to be an effective anti-atherogenic agent. This strategy proved to be highly effective in inhibiting atherosclerosis in animals. We have expanded on this strategy to express other anti-inflammatory genes such as Jmjd-3, SOCS-1, IRF-4 and IL-37 genes to assess their effects on vascular diseases. We are also interested in tackling other conditions that are known to be inflammation-driven such as rheumatoid arthritis using this technology.
5. Role of chitinase in atherosclerosis

Chitinase 1 (CHIT1), a member of glycosyl hydrolase family 18, is secreted by activated macrophages of mammals. Interestingly, chitinase activity is raised in the sera of atherosclerotic patients and is present in atherosclerotic plaque, although the reason for its presence has not been identified. Despite these findings, the physiological role of CHIT1 in the pathogenesis of atherosclerosis is unknown. In our preliminary studies, DNA microarray assay performed with mRNA from atherosclerotic arteries of cynomolgus monkeys revealed CHIT1 expression to be closely correlated with atherosclerosis development and with areas of macrophage infiltration. We currently have mouse models of CHIT1 deficiency and overexpression and we are in the process of elucidating the role that CHIT1 plays in macrophage function and in atherogenesis.
6. Extracellular RNA in macrophage function and atherosclerosis
In a highly collaborative study with my colleagues in Germany we found that extracellular RNA (eRNA) is abundantly present in atherosclerotic lesions of high fat diet-fed low density lipoprotein receptor-deficient (Ldlr-/-) mice in a time-progressive fashion. We also determined that eRNA causes inflammatory changes in mouse primary macrophages including polarization of the cells to the M1 phenotype with up-regulated expression of inflammatory cytokines such as TNF-a, IL-6 and IFN-g. In a model of accelerated atherosclerosis after arterial injury in apolipoprotein E-deficient (apoE-/-) mice, treatment with RNase1 diminished the increased plasma level of eRNA, evidenced after injury. Likewise, RNase1 administration reduced neointima formation compared to vehicle-treated apoE-/- controls, and was associated with a significant decrease in plaque macrophage content. We are interested in investigating how macrophages are affected by eRNA, including how macrophages recognize eRNA and which signaling pathways are elaborated intracellularly upon its recognition.
7. Atherosclerotic cardiovascular diseases in HIV-infected individuals
It is well known that HIV infected patients have a higher rate of cardiovascular diseases, especially atherosclerosis. In collaboration with researchers here at the UH AIDS Center, we have been studying possible mechanisms behind this phenomenon. Our lab has focused on whether monocyte/macrophages that have been infected with HIV may be one of the culprits. We have convincing ex vivo data that HIV-infected macrophages behave very differently than non-infected cells and that this may contribute to the exacerbation of atherosclerosis in infected individuals. In collaboration with a group at the University of North Carolina that specializes in development of humanized mice, we are currently developing an appropriate murine model to test our theory in vivo.
8. Role of SARS-CoV-2-infected macrophages in COVID-19
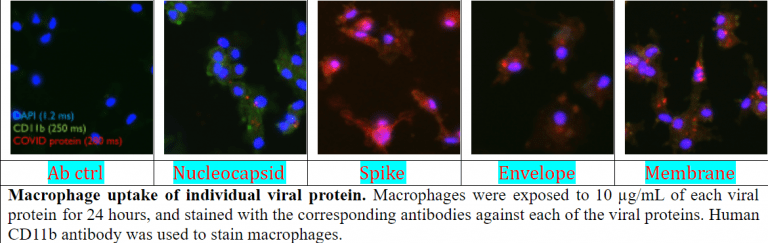
Aside from the cells in the respiratory system, SARS-CoV-2 can infect cells of the immune system which can cause hyperinflammation. This heightened immune response can eventually lead to sepsis (aka cytokine storm) and organ failure. The most important immune cell that contributes to the hyperinflammation in some COVID-19 patients is the macrophage. However, how the SARS-CoV-2 and the small components of the virus that can be found in the tissues and blood of those infected interact with and cause macrophages to become activated is virtually unknown. We are currently investigating how the viral components interact with macrophages, and more importantly, how the macrophage response to the infection can turn into the deadly hyperinflammation in some SARS-CoV-2-infected individuals.
Boisvert WA, Spangenberg J, Curtiss LK. Treatment of severe hypercholesterolemia in apolipoprotein E-deficient mice by bone marrow transplantation. J. Clin. Invest. 1995;96:1118-1124.
Boisvert WA, Spangenberg J, Curtiss LK. The role of leukocyte-specific low density lipoprotein receptors on plasma lipoprotein distribution and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1997;17:340-347.
Boisvert, WA, Santiago, R, Curtiss, LK, Terkeltaub, R. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophage in atherosclerotic lesions of LDL- receptor-deficient mice. J. Clin. Invest. 1998;101:353-363.
Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Nagy L, Edwards PA., Curtiss L.K., Evans R.M. and Tontonoz P. A PPAR-γ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular Cell 2001;7:161-171.
Boisvert, W.A., The participation of inflammatory cells in atherosclerosis. Drugs of Today, 37(3):173-179, 2001.
Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, Curtiss LK, Berliner JA, Boisvert WA. Overexpresion of interlelukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL-receptor deficient mice by altering lymphocyte and macrophage phenotypes. Circ. Res. 2002;90:1064-1071.
Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM. PPARd controls atherogenic inflammation by ligand-directed co-factor switching. Science 2003;302:453-457.
Boisvert WA. Modulation of atherogenesis by chemokines. Trends Cardiovasc. Med. 2004;14:161-165.
Boisvert WA, Rose DM, Johnson KA, Fuentes ME, Lira SA, Curtiss LK, Terkeltaub RA. Upregulated expression of the CXCR2 ligand, KC/GRO, in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am. J. Pathol. 2006;168:1385-1395.
Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, Curtiss LK, Terkeltaub R. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler. Thromb. Vasc. Biol. 2006;26:563-569.
Boisvert WA, Rose DM, Johnson KA, Fuentes ME, Lira SA, Curtiss LK, Terkeltaub RA. Upregulated expression of the CXCR2 ligand, KC/GRO, in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am. J. Pathol. 2006;168:1385-1395.
Wang H, Oyama N, Rikitake Y, Kitamoto S, Gitlin J, Liu P-Y, Liao JK, Boisvert WA. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR-/- mice. FASEB J. 2008;22:3561-3570.
Gitlin JM, Homeister JW, Bulgrien J, Counselman J, Curtiss LK, Lowe JB, Boisvert WA. Disruption of tissue-specific fucosyltransferase VII, an enzyme necessary for selectin ligand synthesis, suppresses atherosclerosis in LDLR-/- mice. Am. J. Pathol. 2009;174:343-350.
Han X, Kitamoto S, Lian Q, Boisvert WA. Interleukin-10 facilitates both cholesterol uptake and efflux in macrophages. J. Biol. Chem. 2009;284:32950-32958.
Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869-2880.